An early clinical sign of DNA damage is muscle weakness. This weakness is a result of reduced production of the energy needed by the muscle in order to contract. Muscles primarily move other parts of the body and this is achieved by a complex contraction system that is initiated by an energy source known as adenosine triphosphate (ATP). In this article we shall discuss the nature of mitochondria, how they supply energy, what causes them to be damaged, and what we can do to help offset this process.
The Mitochondria
The word mitochondria is derived from two German words, meaning thread and corn. Early researchers saw these cells differently, depending on what position the cells were in. They appeared to look like a kernel of corn when viewed on end, but when viewed longitudinally, they appeared to be threadlike. Those early impressions were combined and, today, we call them mitochondria. We are uncertain of the origin of the mitochondria, but we know that it is most likely from a specific type of bacteria that entered our cell somewhere around two million years ago when the atmosphere contained toxic levels of oxygen. Most species died because they were unable to handle oxygen; however, some survived, and if it were not for these mitochondria entering these early cells, we would not be here.
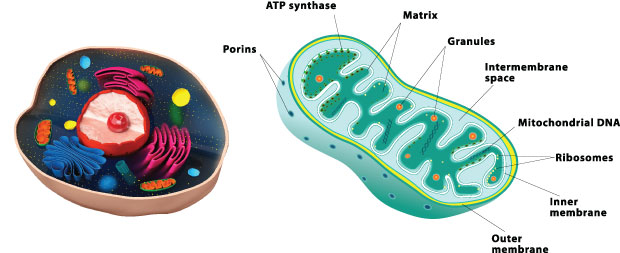
The current teaching about mitochondria is that they function only as “energy factories” for the cell. This is a mistake which has impeded our understanding of the true biology of mitochondrial disease. There are 3,000 genes needed to make a mitochondrion, and mitochondrial DNA encodes only 37 of these genes; the rest of the genes are held in the cell nucleus and the proteins produced are transported to the mitochondria. A mere three percent of the genes necessary to make a mitochondrion (100 of the 3,000) are allocated for making ATP. The other 2,900 of 3,000 genes are involved with specialized duties of the differentiated cell in which the mitochondrion resides. It has been found that these duties change as we develop from embryo to adult, going through the processes of tissues growth, maturing, and environmental adaptation. To name a few of the non-ATP related functions, they are involved with many of the major metabolic pathways in the cell to build, break down, and recycle its molecular building blocks. It is interesting that cells cannot make the ribonucleic acid (RNA) and DNA they need to grow and function without the help of mitochondria. Remember that RNA and DNA are made up of purines and pyrimidines, the essential bases. Mitochondria have the critical enzymes for pyrimidine biosynthesis, dihydroorotate dehydrogenase, and heme synthesis, d-amino levulinic acid synthetase, the major compound required to make hemoglobin. Mitochondria are specialized to detoxify ammonia in the urea cycle in the liver. These little mitochondria are even important for cholesterol metabolism, estrogen and testosterone production, metabolize neurotransmitter, and to detoxify free radical production. Besides these duties, they also break down (oxidize) the fat, protein, and carbohydrates we eat and drink.
Essentially, for our purposes, mitochondria make ATP and our constant source of energy. They do this by extracting energy from the food we eat, reducing the food first to high-energy electrons and then extracting the energy from the electrons, then passing it to a molecular source that can store and then supply the energy as needed. Briefly, this process is as follows.
Mitochondria are organelles with their own genome that consume oxygen and metabolic byproducts of digestion to generate ATP, but at the same time they produce ROS. They have many other cellular functions such as signal transduction, cell cycle regulation, oxidative stress, thermogenesis, and apoptosis. When more energy is needed, they undergo biogenesis, to supply more mitochondria. Mitochondria produce energy from substrates through the tricarboxylic acid (TCA) cycle and the electron transport system (ETS) to generate ATP. The ETS is made up of five complexes (I-V) embedded in the inner mitochondrial membrane (IMM). These complexes receive electrons from reducing equivalents called nicotinamide adeninine dinucleotide reduced (NADH) and flavin adenine dinucleotide reduced (FADH2), which are generated by dehydrogenase activity in the TCA. The electrons are passed along to the complexes with O2 being the final acceptor at complex IV. The propensity to accept an electron increases along the chain of complexes; this creates enough energy to drive hydrogen ions across the IMM. The transfer of hydrogen ions creates a proton gradient and membrane potential that drives the synthesis of ATP as protons flow back to the matrix via complex V (ATP synthase). This process is also known as oxidative phosphorylation, or OXPHOS. The electron transport system has a significant proton “leak” by the movement of hydrogen ions back into the matrix space that is not mediated through complex V. This type of proton transfer can be uncoupled from ATP phosphorylation, which means no energy is produced; this inefficiency contributes to the demand for more reducing equivalents such as NADH.
Since mitochondria can still function, many research workers do not feel that mitochondrial function is impaired and, therefore, is not implicated in cellular disturbances associated with aging and chronic diseases. Actually, the definition of mitochondrial dysfunction itself is the subject of controversy, though many diseases have been identified as being caused by serious mitochondrial dysfunction. Mitochondrial diseases appear to cause the greatest damage to cells composing the brain, heart, liver, skeletal muscles, kidney, and the endocrine and respiratory systems. They may manifest with loss of motor control, muscle weakness and pain, gastro-intestinal disorders and swallowing difficulties, poor growth, cardiac disease, liver disease, diabetes, respiratory complications, seizures, visual/hearing problems, lactic acidosis, developmental delays, and susceptibility to infection, depending on the affected cell. In this article, we will concentrate our interest on how mitochondrial dysfunction affects skeletal muscles which results in aging symptoms. First, we shall look at the impact of aging on the biochemical and bioenergetic pathways in skeletal muscle mitochondria.
In the aging process, mitochondria are described best by changes in oxidative stress, or a decay in mitochondrial DNA, along with a reduction in some enzyme activities and alterations in mitochondrial respiration, resulting in changes of the structure and appearance of mitochondria. In aging, some mitochondrial organelles are enlarged and more rounded in appearance. Inside the mitochondria, shorten cristae and vacuolization of the matrix are seen, and the density of mitochondria in skeletal muscle drops appreciably. The mitochondria constitute the major source of ROS inside the cell, with mitochondrial complexes I and III being the main sites of superoxide generation, contributing the most to reactive oxygen species production. The reactive oxygen species, including O2.- and H2O2, can cause oxidative damage to surrounding structures, including mtDNA, which is highly susceptible to free radical damage. Oxidation by ROS generates faulty proteins, oxidized lipids, and mtDNA mutations, all leading to cellular and mitochondrial dysfunction. These appear to be the major processes implicated in the mitochondrial theory of aging. This increase in ROS production is associated with oxidation of ETS complex V, leading to decreased ATP; thus, a decrease in available energy.
The ETS, being in the inner mitochondrial membrane, is the primary site of ROS production; thus, is the main source of oxidative stress resulting in damage to proteins, lipids, and both DNA in the mitochondria and in the cell. It is known that free radical superoxide anions (O2.-) are generated when electrons are passed from complexes I and III of the ETS go to O2, instead of the appropriate ETS subunit. As much as two to four percent of total oxygen consumption may go toward the production of ROS, rather to create energy as ATP. Enzymes that scavenge these free radical are important mitochondrial defenses against oxidative stress by neutralizing O2.- within the mitochondrial matrix. Enzymes such as superoxide dismutase and SOD2 catalyze the reduction of mitochondrial SOD2-generated H2O2 to nontoxic H2O in the mitochondria and the cell with glutathione peroxidase and catalase. In young muscle, mitochondria are numerous and efficient, but in aging muscle mitochondria are less numerous and appear to become impaired and develop reduced oxidative capacity. Exercise and caloric restriction, or caloric restriction mimetics, can improve impairments in aged muscle. Once thought of as relatively static round organelles, mitochondria are now recognized as highly dynamic, existing in networks that are constantly being remodeled by biogenesis, fusion and fission, and degradative processes such as autophagy. Through these dynamics, they both respond to and drive cellular processes, including apoptosis, whose dysregulation is thought to be a key factor in sarcopenia.
 "Do we decrease physical activity as we age or does it occur because of the inefficiency of our muscular system?"
"Do we decrease physical activity as we age or does it occur because of the inefficiency of our muscular system?"
MITOCHONDRIAL RESPONSES
Biogenesis
Mitochondrial biogenesis is the expansion of existing mitochondrial activity. There are various ways this can occur: through growth of the mitochondrial network which increases mitochondrial mass, or division of preexisting mitochondria which increases mitochondrial number. The trigger is a need for more energy that exceeds respiratory capacity, which happens in response to exercise, stress, hypoxia, nutrient availability, hormones, especially insulin, ROS production, and temperature changes.
When biogenesis is triggered, the nuclear genome produces the mitochondrial regulatory factors, which, after they imported into the mitochondria, initiate replication and transcription of mtDNA, expanding the mitochondrial network. Once the process is triggered, a series of molecular biochemical processes are initiated and ultimately results in the production of more mitochondria. The processes involved are beyond the scope of this article, but in essence they require a cooperative effort between the cell’s nucleus and the DNA of the mitochondria known as mtDNA. For those readers who are interested in the molecular biology, all references are supplied.
Mitochondrial Turnover
Mitochondrial turnover is promoted by the autophagy-lysosome system, a cellular housekeeping system that degrades mitochondria, as well as other cellular components. When mitochondria pass through the autophagy-lysosome system, it is known as “mitophagy.” In mitophagy, dysfunctional mitochondria are recognized and engulfed in a double-membrane structure called a phagophore. After this stage, vesicles called autophagosomes are formed. The autophagosomes then fuse with the lysosome, producing autolysosomes, and the contents are recycled. There is not much known about the role of mitophagy in the aging of skeletal muscle. Evidence suggests that mitophagy selectively removes defective mitochondria that are depolarized or produce excessive ROS. However, suppression of autophagy creates an increased ROS production, and reduced oxygen consumption, which results in higher mtDNA mutation rates. It has been found that as we age, autophagy declines both in general and in the skeletal muscle of aged rats. All this means that reduced autophagy rates may contribute to muscular dysfunction. Mice with lower resting oxygen consumption experience increased oxidative stress and higher rates of apoptosis; these mice also suffer from muscle atrophy, weakness, and myofibril degeneration. Studies in a few species indicate that enhanced autophagy may increase lifespan.
 HOW TO ATTENUATE MITOCHONDRIAL AGING
HOW TO ATTENUATE MITOCHONDRIAL AGING
Exercise
There are many benefits to exercise. Exercise can induce mitochondrial biogenesis, up regulate smooth muscle gene expression, along with additional protein synthesis and at the same time, increase smooth muscle oxidative capacity. The big question is do we decrease physical activity as we age or does it occur because of the inefficiency of our muscular system? There is no question that mitochondrial capacity is reduced in elderly subjects. Studies have found that activity levels are significantly reduced in the elderly group, suggesting that independent of exercise training, simply living an active lifestyle may have a significant impact on mitochondrial function. However, this requires more study and has not been definitively proven. Most elderly individuals exercise less than two hours per week. There is no question that exercise training can improve skeletal muscle mitochondria in elderly individuals. Just four months of aerobic exercise can increase protein synthesis and mitochondrial enzyme activity, as well as the expression of genes involved in mitochondrial action, often to the level of those found in young adults.
Exercise does show a significant increase in the mitochondrial function. There is evidence that some impairment remains. The AIDS associated decline in mitochondrial oxidative capacity can be reduced in older subjects who exercise regularly. However, it does not restore the capacity of mitochondrial proteins mitochondrial DNA content, or other aspects of mitochondrial function. This would indicate that there is a persistent dependent impact on mitochondria. The data so far is relatively short-term ranging from 12 to 24 weeks. The question is what is the impact of active lifestyles when there are significant changes, many going from a sedentary life to one of increased activity? This is good news for those individuals who are close to exercise, but nevertheless remain considerably active.
Caloric Restriction
Caloric restriction (CR) has been given a considerable amount of press recently, but not without reason. It appears to be the most significant anti-aging activity so far developed. It is known to retard primary aging and secondary aging, which is the type of aging due to disease and negative lifestyle. The net result is an increase both in the median and maximum lifespan in many species; there is limited data, however, on primates, but the evidence suggests that reduction in our food consumption by 20 to 40 percent fewer calories will extend lifespan up to 50 percent and reduce the incidence of many age-associated diseases, including both cancer and metabolic disorders.
How does this happen? The benefits ascribed to CR are believed to be due in large part to reductions in oxidative stress. A 10-year study in primates with CR intervention resulted in marked decreases in oxidative damage to lipids and proteins. Caloric restricted animals exhibit fewer mtDNA and nuclear DNA mutations and less oxidative damage to skeletal muscle mitochondria than their regularly fed counterparts. CR appears to positively affect mitochondrial efficiency, content, and function. It does this by lowering energy expenditure in animals and humans, apparently by producing mitochondria that are able to maintain normal levels of ATP, yet consume less oxygen. It is generally believed that this energetic adaptation is mediated via decreased proton leak, which has been confirmed in rodent studies, and that decreased proton leak, in turn, is enabled by the shift to a less oxidative milieu in terms of mitochondrial content and function. CR does not affect the gene expression, protein level, or activity of citrate synthase, nor the activities of other TCA proteins, however, caloric restriction does affect some ETS enzymes. In particular, complex IV activity, which is consistently responsive to caloric restriction shows increased activity in comparison to that in aged rodents and usually similar activity to their younger counterparts.
In a nutshell, food is fuel, and the more fuel you feed into a machine the shorter is its lifespan. A prime example is a furnace, which, if operated at high temperature, has a short life. The same is true for automobiles; compare the life of a race car to that given by a typical suburbanite. It is a matter of simple biology – the more you eat, the shorter your life. Lifespan is inversely proportional to the amount of food we eat.
Caloric Restriction Mimetics
CR is a hard sell. Most individuals would prefer a pill to prevent the need for dieting and exercise. The search is on for something that will mimic CR and it appears to have been found in a plant extract known as resveratrol. How resveratrol affects mitochondria has been well studied, and it is being shown to improve exercise capacity, motor function, and reduce metabolic dysfunction.
References
1 Hipkiss, A.R. “Mitochondrial Dysfunction, Proteotoxicity, and Aging: Causes or Effects, and the Possible Impact of NAD+-controlled Protein Glycation.” Advances in Clinical Chemistry 50 (2010): 123–150.
2 Johannsen, D.L., Conley, K.E., Bajpeyi, S., et al. “Ectopic Lipid Accumulation and Reduced Glucose Tolerance in Elderly Adults Are Accompanied by Altered Skeletal Muscle Mitochondrial Activity.” The Journal of Clinical Endocrinology and Metabolism 97.1(2012): 242–250.
3 Saraste, M. “Oxidative Phosphorylation at the Fin de Siecle.” Science, 283. 5407: 1488–1493.
4 Holloszy, J.O. “Skeletal Muscle, ‘Mitochondrial deficiency’ Does Not Mediate Insulin Resistance.” The American Journal of Clinical Nutrition 89.1 (2009): 463S–466S.
5 Sarcopenia (from Greek σάρξ sarx, “flesh” and πενία penia, “poverty”) is the degenerative loss of skeletal muscle mass (0.5-1% loss per year after the age of 25), quality, and strength associated with aging
6 Kujoth, G.C, Hiona, A., Pugh, T.D., et al. “Medicine: Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging.” Science 309.5733 (2005); 481–484.
7 Autophagy (or autophagocytosis) (from the Greek auto-, “self” and phagein, “to eat”), is the basic catabolic mechanism that involves cell degradation of unnecessary or dysfunctional cellular components through the actions of lysosomes.[1] The breakdown of cellular components can ensure cellular survival during starvation by maintaining cellular energy levels.
8 Holloszy, J.O. “Biochemical Adaptations in Muscle. Effects of Exercise on Mitochondrial Oxygen Uptake and Respiratory Enzyme Activity in Skeletal Muscle.” Journal of Biological Chemistry 242.9, (1967) : 2278–2282.
9 Parise, G., Brose, A. N. & Tarnopolsky, M.A. “Resistance Exercise Training Decreases Oxidative Damage to DNA and Increases Cytochrome Oxidase Activity in Older Adults.” Experimental Gerontology 40.3 (2005): 173–180.
10 Lee, C.M., Aspnes, L. E., Chung, S. S., et al. “Influences of Caloric Restriction on Age-associated Skeletal Muscle Fiber Characteristics and Mitochondrial Changes in Rats and Mice.” Annals of the New York Academy of Sciences 854 (1998): 182–191.
11 Speakman, J.R and S. E. Mitchell, “Caloric Restriction,” Molecular Aspects of Medicine 32.3: 159–221.
12 Usuki, F., Yasutake, A., Umehara, F., and Higuchi, I. “Beneficial Effects of Mild Lifelong Dietary Restriction on Skeletal Muscle: Prevention of Age-related Mitochondrial Damage, Morphological Changes, and Vulnerability to a Chemical Toxin.” Acta Neuropathologica 108.1 (2011): 1–9.
13 Lagouge, M., Argmann, C., Gerhart-Hines, Z., et al., “Resveratrol Improves Mitochondrial Function and Protects Against Metabolic Disease by Activating SIRT1 and PGC-1α,” Cell 127. 6 (2006): 1109–1122.
14 Murase, T., Haramizu, S., Ota, N., and Hase, T. “Suppression of the Aging-associated Decline in Physical Performance by a Combination of Resveratrol Intake and Habitual Exercise in Senescence-accelerated Mice.” Biogerontology 10.4 (2009): 423–434.
 Michael Q. Pugliese, BS became the third-generation CEO of Circadia by Dr. Pugliese, Inc. in 2006. Under his leadership, the Circadia brand has grown to achieve international recognition and distribution. Pugliese is a licensed aesthetician, a member of the Society of Cosmetic Chemists, and regularly attends their education events to stay on the cutting edge of new product development. His compelling original lectures honor the tenets of modern skin science discovered by his grandfather, and add today’s application of that information in an ever-changing business and scientific environment.
Michael Q. Pugliese, BS became the third-generation CEO of Circadia by Dr. Pugliese, Inc. in 2006. Under his leadership, the Circadia brand has grown to achieve international recognition and distribution. Pugliese is a licensed aesthetician, a member of the Society of Cosmetic Chemists, and regularly attends their education events to stay on the cutting edge of new product development. His compelling original lectures honor the tenets of modern skin science discovered by his grandfather, and add today’s application of that information in an ever-changing business and scientific environment.